Abstract and Introduction
Abstract
Reading facial emotion is disrupted by both psychopathology, such as autism, and altered function of neurotransmitter, such as serotonin. These effects could result from reduced sensitivity of emotional processing systems to facial emotion. The impact of facial expression is also greater when personally directed than when averted. We therefore hypothesized that brain activity associated with emotional representation, would be more susceptible to manipulation of serotonin function by Acute Tryptophan Depletion (ATD) for front-viewed than side-viewed faces, measured using functional imaging (fMRI). ATD reduced activity independent of face view in left superior temporal sulcus (STS) and anterior cingulate. In temporal pole, medial frontal cortex and orbitofrontal cortex, ATD also reduced activity, but specifically for front-viewed faces. In right STS, ATD increased activity, but specifically for side-viewed faces. Activity in the amygdalae depended on face view and emotion type. We suggest that engagement of empathic and associative learning functions when viewing faces is facilitated by direct facial view and intact serotonin transmission. Averted faces, and reduced serotonin function facilitate attention to the external goal of gaze. These changes could be adaptive in a threatening context and markedly affect empathic function in conditions associated with impaired serotonin function, such as depression and autism.
Introduction
The judgment of emotional expression is an everyday cognitive task that may have important effects on the ability to maintain healthy social interaction, and it has been shown to be adversely affected by a range of psychopathology. Individuals with autism show impaired ability to recognize emotion (Baron-Cohen et al., 1999) and those with Turner's syndrome appear to be deficient in recognizing fear (Lawrence et al., 2003). People who are alcohol dependent are more likely to perceive sad faces as hostile (Frigerio et al., 2002) whilst psychopaths show poor ability to recognize fear (Blair et al., 2004). Mania, depression and schizophrenia have also been related to impaired processing of facial emotion (Rubinow and Post, 1992; Lembke and Ketter, 2002; Bediou et al., 2005).
Facial emotion may be judged through learned associations. Associative conditioning processes are served by a network that includes amygdala and orbitofrontal cortex (OFC). The amygdala responds quickly to emotionally potent stimuli and assigns importance to them (Phelps, 2006). The (OFC) has close connectivity with the amygdala but also with other cortical and subcortical structures that give it an important role in learning, particularly in relation to facial expression (Blair et al., 1999; Kringelbach and Rolls, 2004).
Expressions may be judged empathically (i.e. by feeling what others feel). While such empathic responses may also be established through associative learning, the phenomenological experience will be qualitatively distinct from other learned associations. Moreover empathic judgments or responses appear to involve distinct brain systems. Empathy appears to rely on structures that integrate sensory information processed in limbic structures with visual information. Singer et al. (2004) found empathy for pain to be associated with activity in caudal anterior cingulate, insula and medial prefrontal cortex (MPFC), whilst other studies have consistently implicated temporal poles (TP) and insulae (Phillips et al., 1997; Lane et al., 1998; Carr et al., 2003; Frith and Frith, 2003; Kim et al., 2005; Vollm et al., 2006). Premotor function associated with generation of 'contagious' motor activity may also be a feature of empathy (Hietanen et al., 1998; Surakka and Hietanen 1998; Gallese et al., 2004).
Either way, whether emotional expression is read through empathy or associative learning, both processes depend upon the emotional intensity of the expression (Morris et al., 1996; Calder et al., 1997; Phillips et al., 1997; Young et al., 1997). More exaggerated emotional expressions are easier to interpret than subtle ones, and have greater effects on emotional perception. Another important variable that mediates the impact of facial expressions is whether the face or gaze is directed to, or averted from, the observer. Gaze direction has well-documented effects on activity of the amygdala (Adams et al., 2003) and amygdala activity may be necessary for attention to be directed to another individual's gaze (Adolphs et al., 2005). Expressions of anger and joy are quicker to recognize when they are direct compared to when averted, though expressions of fear or sadness are quicker to recognize when averted (Adams and Kleck, 2003). Gaze-direction may also influence recruitment of other brain areas to emotion processing. Wicker et al. (2003) found direct gaze promoted recruitment of superior temporal gyrus when participants were asked to judge if a face was friendly or hostile. Face as well as gaze direction may also be important but has been less explored. Sato et al. (2004) found that a direct view of a face (seeing it 'face-on'), increased recruitment of the left amygdala to emotional judgment.
Just as variation in the intensity of the expression may affect its impact, so may variation in the observer's sensitivity to it. This may be the result of psychopathology affecting emotional judgment through differential processing of direct and averted gaze. Dalton et al. (2005) found that individuals with autism spent less time looking at the eyes when judging emotion, and that fusiform and amygdala activity correlated with time spent looking at the eyes. Directed facial expression in communication is a discriminative diagnostic feature of autism, suggesting that perceiving gaze direction may influence behavioral development (Lord et al., 2000; Williams, in press). Observer sensitivity may also be affected by serotonin (5-HT) function. Acute administration of tryptophan (the dietary precursor of serotonin) is associated with improved emotion recognition (Attenburrow et al., 2003), whilst acute tryptophan depletion (ATD) is associated with increased errors (Harmer et al., 2003). The molecular mechanisms underlying these processes are unclear. A simple explanation is that tryptophan excess stimulates serotonin synthesis whilst deprivation prevents it. This simple mechanism has been questioned and it has since been suggested that effects of ATD may be mediated through effects on 5-HT2 receptors rather than direct effects on extracellular 5-HT concentrations (Yatham et al., 2001). Nevertheless, it seems likely that ATD reduces serotonin function.
Serotonin function may facilitate emotional processing through empathic and associative learning functions. Serotonergic projections are widespread in the cortex, and impact upon limbic and higher cognitive functions in prefrontal and limbic regions (Canli et al., 2005; Robbins, 2005; Evers et al., 2006). The anterior cingulate region, associated with empathy and emotion processing (Lane et al., 1998; Singer et al., 2004; Amodio and Frith, 2006), receives extensive serotonergic input (Celada et al., 2001). In individuals with depression, selective serotonin reuptake inhibitors (SSRIs) affect blood flow to this region (Kennedy et al., 2001). Serotonin appears to be important for facial processing in the amygdala. SSRIs modify processing of threat in the amygdala (Cools, 2005; Harmer et al., 2006), and individuals with genetically determined, lower serotonin function show reduced amygdala activity when processing facial expressions (Hariri et al., 2002).
The purpose of this study was to further characterize the role of serotonin function in emotional processing by manipulating serotonin function during fMRI using ATD. We hypothesized that faces viewed from the front, would engage emotional processing systems more powerfully than those viewed from the side, and consequently would be more susceptible to manipulation of serotonergic function. We predicted that activity in anterior cingulate, temporal pole and insula would reflect empathic function and the amygdala and OFC would reflect emotional learning. In these systems we expected to find that emotional expressions directed at the observer would be more affected by ATD than emotional expressions directed away from the observer. We also predicted modulation according to type of emotion. For example, fear processing would be processed preferentially by the amygdala (Calder et al., 1996; Morris et al., 1996), and serotonin depletion would have greater effects on fear.
Methods
Participants and General Procedures
The study was approved by the Grampian University Hospitals Ethical Committee. Individuals were recruited through advertisements. All participants were right-handed males aged between 18 and 30 years. A consultant psychiatrist interviewed all participants prior to participation to exclude a history of depression or current depression, neurological illness or disability. Thirteen individuals commenced the experiment. Two were unable to complete the second stage following adverse emetic responses to the amino-acid mixture, and a further one was later removed from analysis due to head movement artifact.
ATD was carried out in a placebo-controlled, double-blind fashion. The active drink contained 100 g of amino acids, made up according to the recipe of Young et al. (1985), and mixed with blackcurrant juice. Each individual participated in the same experiment twice, at intervals of approximately 2 weeks. In the active tryptophan depleting condition (T-), the drink contained no tryptophan. In the control condition (T+), tryptophan was added to the drink. Serum tryptophan was assayed at time of taking the drinks and at time of scanning. On each occasion, participants presented to the Department of Child Health at 9:00 am having fasted from the previous midnight. A blood sample was taken and the amino-acid drink was administered. Five hours later participants attended the scanning suite whether a further venous blood sample was taken before scanning commenced.
Experimental Task
Participants were shown an image of a static face (see Figure 1 for example) and asked to choose whether each facial image was neutral or emotional in expression, responding on a key-pad. Ten different stimuli types were shown as in Table 1 (and illustrated in Figure 1.) Stimuli were original photographs masked to remove all features extraneous to the face, and showed either a neutral, fearful or happy expression. The level of expression was set relatively high, at either 50% or 100% of full expression to minimize performance differences that would confound interpretation, but also to make the task sufficiently demanding of attention.

Figure 1.
Examples of Face-stimuli; Fear Front 100%; Fear Side 100%; Happy Front 50%; Happy Side 100%; Neutral Front; Happy Side 50%.
There were five conditions: Happy-front, Happy-side, Fear-front, Fear-side and baseline. Stimuli were presented in the ratio of one-third 100% emotional expression, one-third 50% emotional expression and one-third neutral. Subjects were asked to decide if the emotional expression was neutral or emotional and then to press the appropriate button on their key-pad. In the baseline condition a cross was presented on the left or right of the screen and subjects were asked to press a button if the cross was on either the left or right of the screen.
Each condition consisted of 12 images in the proportions above, each presented for 2.5 s. Total block time was 32.5 s including 2.5 s for instruction. Each condition was repeated four times for a total session length of 10 min 50 s (5 conditions X 4 repeats X 32.5 sec). The session was repeated twice.
Scanning Method and Analysis
Functional imaging (FMRI) was performed using a 1.5 T scanner (NVi, General Electric Medical Systems, Milwaukee, WI). A quadrature head coil was used to obtain high-resolution gradient echo 3D volumetric images and four sets of functional images using blood oxygenation level dependent contrast. The high-resolution images were collected using a T1 weighted sequence with the following parameters: field of view, 24 cm; 20/6, (TR/TE); flip angle, 35°; slices, 124; slice thickness, 1.6 mm; and matrix, 256 X 256. FMR images were acquired in axial planes with a {T }2 *-weighted single shot, gradient-echo, echo-planar pulse sequence with the following parameters: field of view, 24 cm; 3000/33, (TR/TE); flip angle, 90°, slices, 24; slice thickness, 5 mm; and matrix, 128 X 128. The head was firmly stabilized between two foam pads.
Data analysis was performed in a two-stage mixed-effects analysis (equivalent to a random effects analysis) in which BOLD responses for each subject were first modeled using a standard hemodynamic function in the context of the fixed-effects general linear model. Subject-specific linear contrasts on the parameter estimates were then entered into a second-level analysis to perform within group analysis using a one-sample t-test, resulting in a t statistic for each voxel. For within group analyses, contrast weights were used to identify clusters of activation. The term 'activation' is defined as voxels showing significantly better fit of the hemodynamic model in one condition compared to another. For direct group comparison, two different weight combinations were used to pursue two kinds of contrast: T- group + 1/T+ group - 1 (to identify voxels showing significantly better fit of the hemodynamic model (i.e. 'greater activation') in the T- than in the T+ group); T+ group - 1/T- group + 1 (to identify voxels showing better fit for the T+ group).
Plasma Tryptophan Measures
The tryptophan assays were performed on the Biochrom 20 Plus amino acid analyser (from Biochrom Ltd, Cambridge, UK). This is a continuous flow liquid chromatography system utilizing cation exchange resin followed by treatment with ninhydrin solution. Plasma, treated with equal volumes of 0.1M HCl and 500 µmol/l norleucine in 10%(w/v) 5-sulphosalicylic acid, was mixed and centrifuged. The supernatant was stored frozen until a batch was collected. The thawed supernatants were treated with 0.50M LiOH and loaded onto the amino acid analyzer. Comparison to standard solutions allowed calculation of the tryptophan concentration.
Results
For each individual, sensitivity to ATD was calculated by measuring the plasma tryptophan level during the active session as a percentage of baseline level. Serum tryptophan was successfully lowered in all participants by up to 92% but the range was considerable (Supplementary Table 1 ). For two individuals, levels during the active session remained at 50-60% of baseline. Initial, group-wise, random effects analyses failed to reveal effects of tryptophan depletion on brain activity. After regression analysis (see subsequently) indicated a dose-dependent response to ATD, we deemed these two individuals to be non-responders and re-ran group-wise analyses of effects of ATD with them excluded. This meant that fMRI data was available from 10 participants (20 sessions) for fMRI analyses concerning the relationships between facial posture and emotional expression, and from eight participants for examining the interactions with tryptophan depletion. Reaction-time data was available for nine participants.
Main Effects
ATD. Our strategy was first to examine the main effects of ATD on whole brain analyses and then to explore interactions and regions of interest. We looked at overall effect of ATD on task by contrasting the sum of all emotional judgment conditions with baseline in the T+ and T- conditions separately, and then looking at where T+ activity was greater than T-. This revealed a focus of activity in left posterior superior temporal sulcus (STS) in addition to other activity. (Figure 2 and Supplementary Table 2 .)
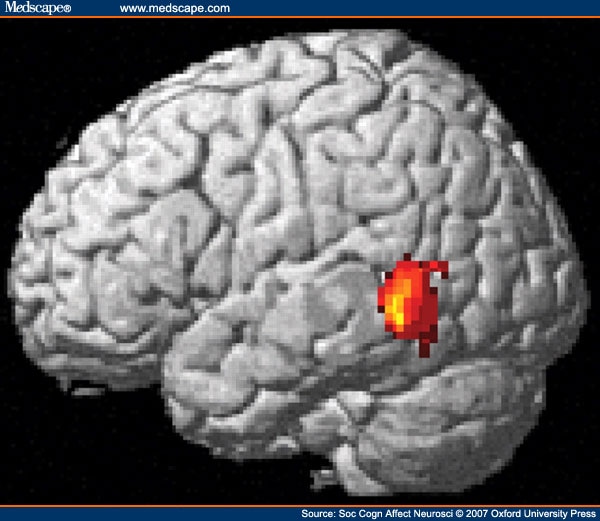
Figure 2.
Main Effects of ATD on Task-related Activity in Left Posterior Superior Temporal Sulcus.
Face direction. Frontal viewing of faces was associated with higher activity compared to side-viewing in visual cortex, cingulate gyrus, premotor cortex and medial OFC (see Supplementary Material: Table 2 and Figure 1).
Emotion type. There were no main effects of emotion at the whole brain level.
Interactions
Emotion type and face-direction. An interaction between emotion and direction of expression was evident in a main cluster in the right parahippocampal gyrus and additional clusters in visual cortex (Supplementary Table 4).
ATD and face-direction. Interactions between ATD and the front-side contrast are shown in Figure 3 and Table 2 . There were five main areas: bilateral STS, the right TP, MPFC, left lateral OFC/dorsolateral prefrontal cortex and left amygdala. These clusters remained significant after correction for multiple comparisons.
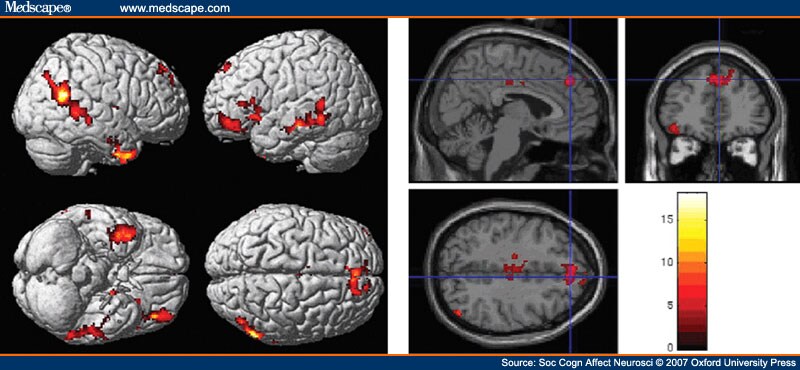
Figure 3.
Interaction between the contrast of front vs side view of face and tryptophan non-depletion vs depletion. Top panel: rendering on cortical surface; Bottom panel: saggital section showing medial prefrontal cortex activity. Main foci are: bilateral along the length of the superior temporal sulcus, the right temporal pole, paracingulate region, left inferior frontal gyrus.
Local Analyses
To ascertain the changes that were driving these interactions, region of interest (ROI) analyses were conducted. ß-values were extracted for cubes of 27 voxels centered on peak voxels in clusters of interest. ATD had a marked impact on activity related to faces viewed from the front in MPFC, rTP, and OFC. Activity in these areas during side viewing of faces changed less with ATD than with front viewing of faces. For right STS and left Broca's area, effects of ATD were greater on side-viewing than front-viewing of faces. Activity that was greater for front compared to side before tryptophan depletion became greater for side compared to front after depletion. Interactions between ATD and emotion type were evident in the amygdala in whole brain analyses, but ROI analysis confined to the STS, MPFC and TPs failed to reveal an interaction between ATD and emotion in these areas. ROI analysis (Figure 4) in the amygdala shows that the interaction between ATD and emotion (x = -22, y = -1, z = -20: P = 0.036; x = 18, y = -7, z = -15: P = 0.040 following small volume correction) was driven through effects on happy as well as fear expressions. In the right amygdala, ATD led the amygdala to respond more strongly to side-happy expressions but had little effect on the response to fear. In the left amygdala, ATD affected the response to face-on-fear but not the response to happiness.
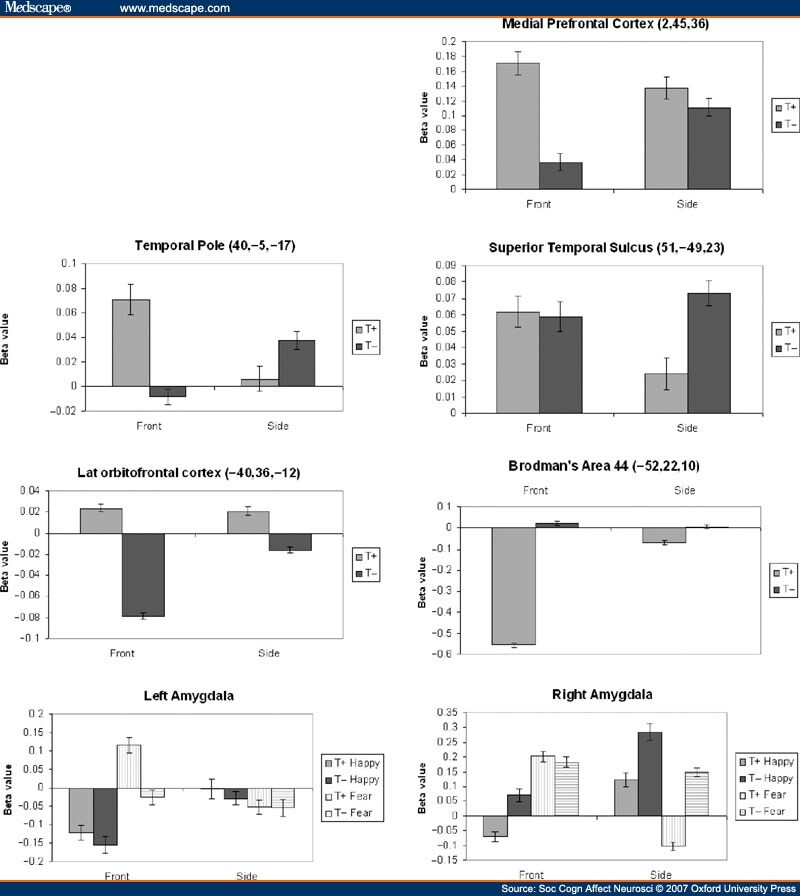
Figure 4.
Graphs showing directions of change in activity following tryptophan depletion, driving interactions shown in Figure 1. Beta-values for 27 voxels surrounding peak value within each cluster, and reflect activity relative to average activity in that area over the duration of scans. Error bars constitute 95% confidence intervals.
Regression Analyses
We predicted individual variation in sensitivity to the ATD protocol, and hypothesized that serotonin function would be disrupted in a dose-dependent fashion in line with the drop in plasma levels of tryptophan. Therefore, in brain areas where serotonin production controlled activity levels, ATD sensitivity would correlate with the BOLD signal. We therefore performed a regression analysis to identify voxels where the mean differences for parameter estimates between active and baseline conditions correlated with plasma tryptophan reduction. We conducted analyses for all conditions together and then emotion types and face direction types separately. Results are shown in Figure 5, and Supplementary Tables 5-7. For all conditions, the largest clusters of correlated activity were located in hippocampus, and the intraparietal sulcus, extending from the angular gyrus along to the superior parietal lobule and precuneus. There were also significant clusters located in the left insula, caudal part of the anterior cingulate, left superior temporal gyrus (STG), and bilateral posterior temporo-occipital junctions. When analyses were separated according to emotion, the extent of depletion correlated more with activity for happy related expressions in anterior cingulate cortex. No correlations were found when analysis was carried out for separate facial directions.
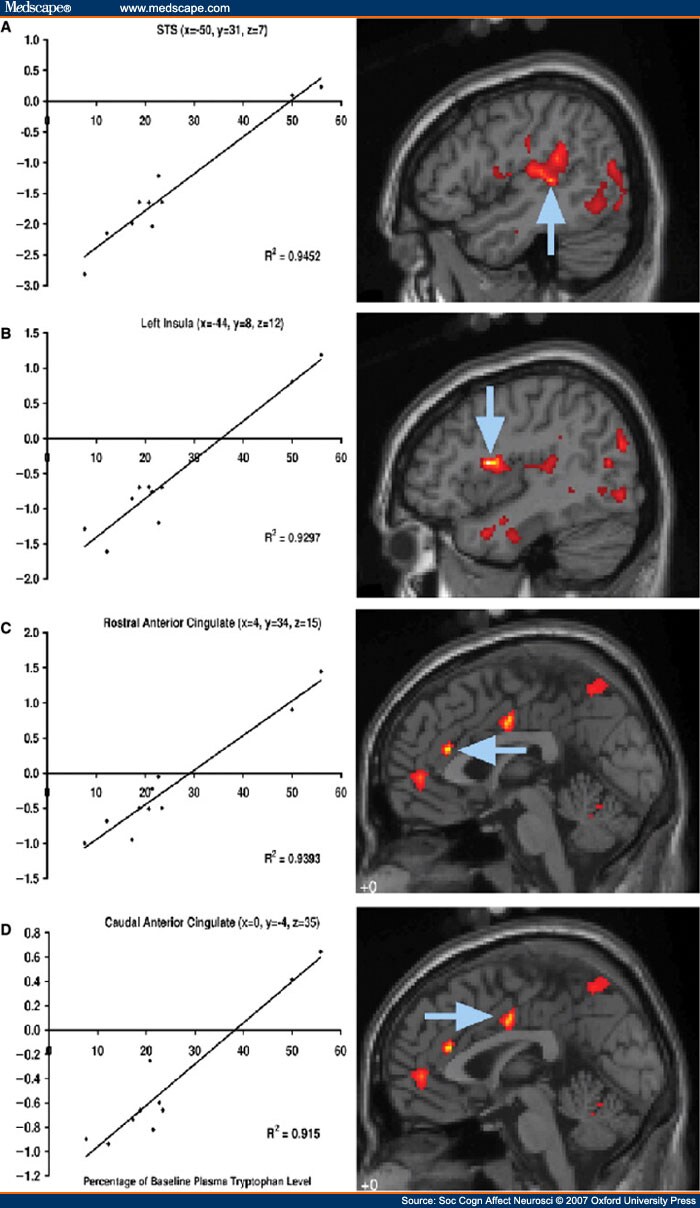
Figure 5.
Areas of cortex where activity correlates strongly with sensitivity to tryptophan depletion. Graphs show serum tryptophan levels at scanning as percentage of baseline levels, plotted against activity averaged over whole cluster during viewing of faces, compared to baseline. Arrows point to peak of cluster. (A) All faces, peak z-score = 4.32, (B) All faces, peak z-score = 5.03 (C) Happy only, peak z-score = 5.15 (D) Happy only, peak z-score = 4.49. Full list of areas affected in supplementary tables 5–7.
Reaction Times
Overall, ATD was associated with a significantly faster response time (Response Time Mean ± 1SE: T- = 1.186 ± 0.076 T+ = 1.311 ± 0.068 s ANOVA main effect of ATD F1,8, = 8.6, P = 0.02). This is unlikely to be the result of a speed/accuracy trade-off because it was not associated with significantly greater numbers of errors (accuracy by mean errors ±1SE: T- = 20.2% ± 3.2% T+ = 14.1 ± 1.5%; ANOVA main effect of ATD non-significant F1,8, = 2.83, P = 0.13). However, ATD was associated with more errors when processing specifically emotions shown in profile, suggesting that ATD may have confounded the greater difficulty of judging emotions seen in profile (Accuracy by mean errors ± 1SE: T-front = 12.5% ± 3.8%, T+ front = 10.2 ± 1.3% T-side = 27.9% ± 3.3% T+ side = 18.0 ± 2.1%; ANOVA interaction between TDP and head view on accuracy judging fear and happiness: F1,8, = 7.89, P = 0.023).
Discussion
We predicted that associative learning and empathic functions serving emotional judgment would be more engaged when facial expression was directed to the observer, and so would be more susceptible to serotonin depletion. The interaction between ATD and facial direction provided support for this hypothesis. Clusters in lateral OFC and amygdala, areas associated with associative learning functions, showed that effects here were much more marked for front-viewed compared to side-viewed expressions. The same contrast also identified clusters in medial frontal cortex (MFC), temporal pole, insula and cingulate gyrus, as well as a dorsal prefrontal cluster that involved Broca's area. The cluster in the MFC maps closely onto an area associated with 'person-perception' (Iacoboni et al., 2004; Mason et al., 2004; Mitchell et al., 2005 Amodio and Frith, 2006) and falls at the boundary between anterior rostral MFC, thought to serve more emotional functions, and posterior rostral MFC thought to serve more cognitive functions (Steele and Lawrie, 2004). This might therefore justify the argument that the front-side X ATD interaction affected only emotional perception. However, areas associated with empathy were also identified in this interaction, including temporal pole (Carr et al., 2003; Frith and Frith, 2003; Kim et al., 2005; Amodio and Frith, 2006; Vollm et al., 2006), caudal cingulate (Lane et al., 1998; Singer et al., 2004), Broca's area and insula (Carr et al., 2003). Therefore, the interaction between ATD and direction of emotional expression is most consistent with an important role for serotonin in mediating the effects of an emotional expression on both associative learning and empathic function, which itself is dependent upon the face being viewed from the front. The regression analysis was also consistent with effects on empathic function, revealing effects of serotonin depletion on caudal anterior cingulate, though these were specific to processing happiness. Correlations may be greater for happiness than fear because of close connectivity with the amygdala, which could contribute more significantly to variance for fear, thus reducing the strength of the correlation. Reaction times were also consistent with effects of ATD on frontal lobe function. ATD resulted in faster response times in the absence of decreased accuracy for front-viewed faces, indicating that it favored more rapid decision making through diminished recruitment of higher cortical function to the task. The response time pattern became more complex when considering side vs front, presumably because of the greater difficulty of judging emotion at a lower impact for side-viewed faces.
Activity in STS was also affected bilaterally by ATD. On the left, this was independent of face view, whereas on the right, activity was increased for side-viewed expressions. Right STS may be more associated with attribution of intention than left STS (Saxe et al., 2004), suggesting that effects of ATD on right STS reflect decreased attention to emotional expression, but increased attention to the goal of gaze. In support of this view, we also found an interaction in BA 44, which is concerned with attributing the relationship between an action and its goal (Kakei et al., 2001; Ochiai et al., 2005). Effects in the amygdala were also lateralized and largely reflected changes in STS. On the right side, ATD increased activity for side-viewed faces, whilst on the left it was reduced or unchanged. There has been some debate with regards to the function of amygdala laterality. Here we suggest that the left is concerned with attention to gaze, whilst the right is more concerned with the goal of the gaze.
The adaptive function of these changes may be related to functions of serotonin in depression and anxiety. As mentioned earlier, it seems likely that ATD reduces serotonergic function, though there may be some question as to the exact mechanism by which this occurs. Reduced serotonin function is associated with greater anxiety and lowered mood, which may be associated with a more threatening and risky environment. In such environments it may be more adaptive to attend to external precipitants of emotional expressions, rather than the underlying mental states. In such cases, attention to the goal of gaze would be more adaptive than attention to the mental state underlying an emotional expression.
A limitation of this study was the low number of subjects in the final analysis of ATD effects. Nevertheless, our approach was robust, using a random effects analysis with a threshold set at P < 0.05 after adjustment for multiple comparisons. Our analyses are unlikely to have resulted in chance findings, though smaller effects may have gone undetected. Including our two non-responders in our regression analyses led to identification of close correlations between extent of tryptophan depletion and activity in anterior cingulate and paracingulate, as well as insula regions and left STS. Correlations are still evident in these regions with the non-responders removed but would not reach the level of significance required to emerge from a whole brain analysis.
To summarize our findings, we suggest that serotonin mediates the impact of a facial expression on emotion perception with marked consequences for empathic reactivity and emotional learning. Impaired serotonin function facilitates a shift of observer's attention to the goal of gaze rather than mental state associated with another's emotional expression. This may reflect adaptive changes to environmental modulators of mood, and may have implications for our understanding of disorders that involve dysfunctional serotonergic activity including depression (Caspi et al., 2003), autism (Devlin et al., 2005; McDougle et al., 2005) and psychopathy (Dolan and Anderson, 2003). Overall our work suggests a potentially powerful neurotransmitter based brain mechanism that could mediate between mental well-being and patterns of thinking that may heavily influence social behavior.
Table 1. Details of Stimuli Types
Front
Side
50%
100%
50%
100%
Neutral
Neutral-front
Neutral-side
Happy
Happy-front (50%)
Happy-front (100%)
Happy-side (50%)
Happy-side (100%)
Fear
Fear-front (50%)
Fear- front (100%)
Fear-side (50%)
Fear-side (100%)
Table 2. Detail of Clusters Shown in Figure 2. Paired T+ vs T-, Interaction With Front vs Side, Responders to ATD Only (n = 8). Loci Reported are Significant at 0.05 Level Corrected for Multiple Comparisons at Cluster Level (Location of Peak of Cluster in Bold, Other Loci are Sub-peaks Within Cluster, Reported to Indicate Direction of Clusters)
X
Y
Z
Hemisphere
Region
BA
Z
Extent
40
-5
-17
Right
Temporal Pole
20
5.08
717
36
-2
-35
Right
Inferior Temporal Gyrus
20
3.84
40
1
-24
Right
Middle Temporal Gyrus
21
3.78
-18
-6
-13
Left
Amygdala
-
4.45
781
-42
-10
-15
Left
Sub-Gyral
20
4.34
-57
-22
-11
Left
Middle Temporal Gyrus
21
4.13
51
-49
23
Right
Supramarginal Gyrus
40
4.42
1004
40
-54
17
Right
Superior Temporal Gyrus
22
4.07
48
-66
40
Right
Inferior Parietal Lobule
39
4.00
2
45
36
Right
Medial Prefrontal Cortex
6
4.12
488
18
49
42
Right
Superior Frontal Gyrus
8
4.01
-8
34
28
Left
Cingulate Gyrus
32
3.53
-28
7
-10
Left
Subcallosal Gyrus
34
4.11
303
-50
22
10
Left
Inferior Frontal Gyrus
45
3.72
-36
19
-6
Left
Inferior Frontal Gyrus
47
3.05
-40
36
-12
Left
Middle Frontal Gyrus
11
3.67
273
-38
48
-16
Left
Middle Frontal Gyrus
11
3.11
-44
52
-6
Left
Middle Frontal Gyrus
10
3.09
-12
-6
44
Left
Cingulate Gyrus
24
3.65
283
-16
-12
36
Left
Cingulate Gyrus
24
2.94
-4
-10
39
Left
Cingulate Gyrus
24
2.89
-10
-39
2
Left
Parahippocampal Gyrus
30
3.61
272
-8
-39
-5
Left
Parahippocampal Gyrus
30
3.11
42
-7
11
Right
Insula
13
3.56
299
40
0
6
Right
Insula
13
3.34
28
-1
13
Right
Lentiform Nucleus
-
3.19
References
|
Acknowledgements
We are grateful to our volunteers, Sophia Durrani for stimulus preparation, and Bill Mutch in the Biochemistry Department of Aberdeen Royal Infirmary for tryptophan measures.
Funding Information
The project was funded by the National Alliance for Autism Research.
Reprint Address
Correspondence to: Dr Justin H. G. Williams, Department of Child Health, University of Aberdeen Medical School, Royal Aberdeen Children's Hospital, Aberdeen. AB25 2ZG. E-mail: justin.williams@abdn.ac.uk
*Department of Child Health, University of Aberdeen Medical School, Aberdeen, Scotland, UK,
1School of Psychology, University of St Andrews, St Andrews, Scotland, UK,
2Department of Radiology, University of Aberdeen, Aberdeen, Scotland, UK, and
3School of Psychology, University of Central Lancashire, Preston, England, UK
Tags: Tryptophan, Emotion
Powered by Qumana